How to represent a protein sequence
September 27, 2023
WIP...
In the last decade, next generation sequencing propelled biology into a new information age. This came with a happy conundrum: we now have many orders of magnitude more protein sequence data than structural or functional data. We uncovered massive tomes written in nature's language, the blueprint of our wondrous biological tapestry, from the budding of a seed to the beating of a heart. But more often than not, we lack the ability to understand them.
An important piece of the puzzle is the ability to predict the structure and function of a protein from its sequence.
Thinking about this as a machine learning problem, structural or functional data are labels. With access to many sequences and their corresponding labels, we can show them to our model and iteratively correct it's predictions based on how closely they match the true labels. This approach is called supervised learning.
When labels are rare, as in our case with proteins, we need to resort to more unsupervised approaches like this:
-
Come up with a vector representation of the protein sequence that captures its important features. The vectors are called contextualized embeddings (we'll refer to them simply as embedding vectors). This is no easy task: it's where the heavy lifting happens and will be the subject of this article.
-
Use the vector representation as input to some supervised learning model. The informative embedding has hopefully made this easier that 1) we don't need as much labeled data and 2) the model we use can be simpler, such as linear or logistic regression.
This is sometimes called transfer learning: the knowledge learned by the representation (1.) is later transferred to a supervised task (2.).
What about MSAs?
We talked in a previous post about ways to leverage the rich information hidden in Multiple Sequence Alignments (MSAs) – the co-evolutionary data of proteins – to predict structure and function. That problem is easier:
However, those solutions don't work well for proteins that are rare in nature or designed de novo for which we don't have enough co-evolutionary data to make a good MSA.
In those cases, can we still make reasonable predictions based on a single amino acid sequence? Another way to look at the techniques in this article is that they are answers to that question. They pick up where MSAs fail. Moreover, models that don't rely on MSAs aren't limited to a single protein family: they understand some fundamental properties of all proteins. Our goal is to build such a model.
Representation learning
The general problem of converting some data into a vector representation is called representation learning, a key technique in natural language processing (NLP). Let's see how it can be applied to proteins.
We want a function that takes an amino acid sequence and outputs embedding vectors. This function is often called an encoder.
Tokens
In NLP lingo, each amino acid is a token. Like amino acid sequences, we can embed an English sentence in the same way, using characters as tokens.
As an aside, words are another reasonable choice for tokens in natural language.
Current state-of-the-art language models use something in-between the two: sub-word tokens. tiktoken is the tokenizer used by OpenAI to break sentences down into lists of sub-word tokens.
Context matters
If you are familiar with earlier NLP embedding models like word2vec, our contextualized embedding is slightly different.
TODO:
Two identical amino acids don't necessarily have the same embedding vector. This is because the embedding vector for each amino acid incorporates context from its surrounding amino acids.
If we want one vector that describes the the entire sequence – instead of a vector for each amino acid – we can simply average the values in each vector.
Now let's create these embedding vectors.
Creating a task
Remember, we are constructing these vectors purely from sequences in an unsupervised setting. Without labels, how do we even know if our representation is any good? It would be nice to have some task: an objective that our model can work towards, along with a scoring function telling us how it's doing.
Let's come up with a task: given the sequence with some random positions masked away
which amino acids should go in the masked positions?
We know the ground truth label from the original sequence, which we can use to guide the model like we would in supervised learning. Presumably, if our model becomes good at predicting the masked amino acids, it must have learned something meaningful about the intricate dynamics within the protein.
This lets us take advantage of the wealth of known sequences (from publicly available large databases such as the Protein Data Bank (PDB)), each of which is now a labeled training example. In NLP, this approach is called masked language modelling (MLM), a form of self-supervised learning.
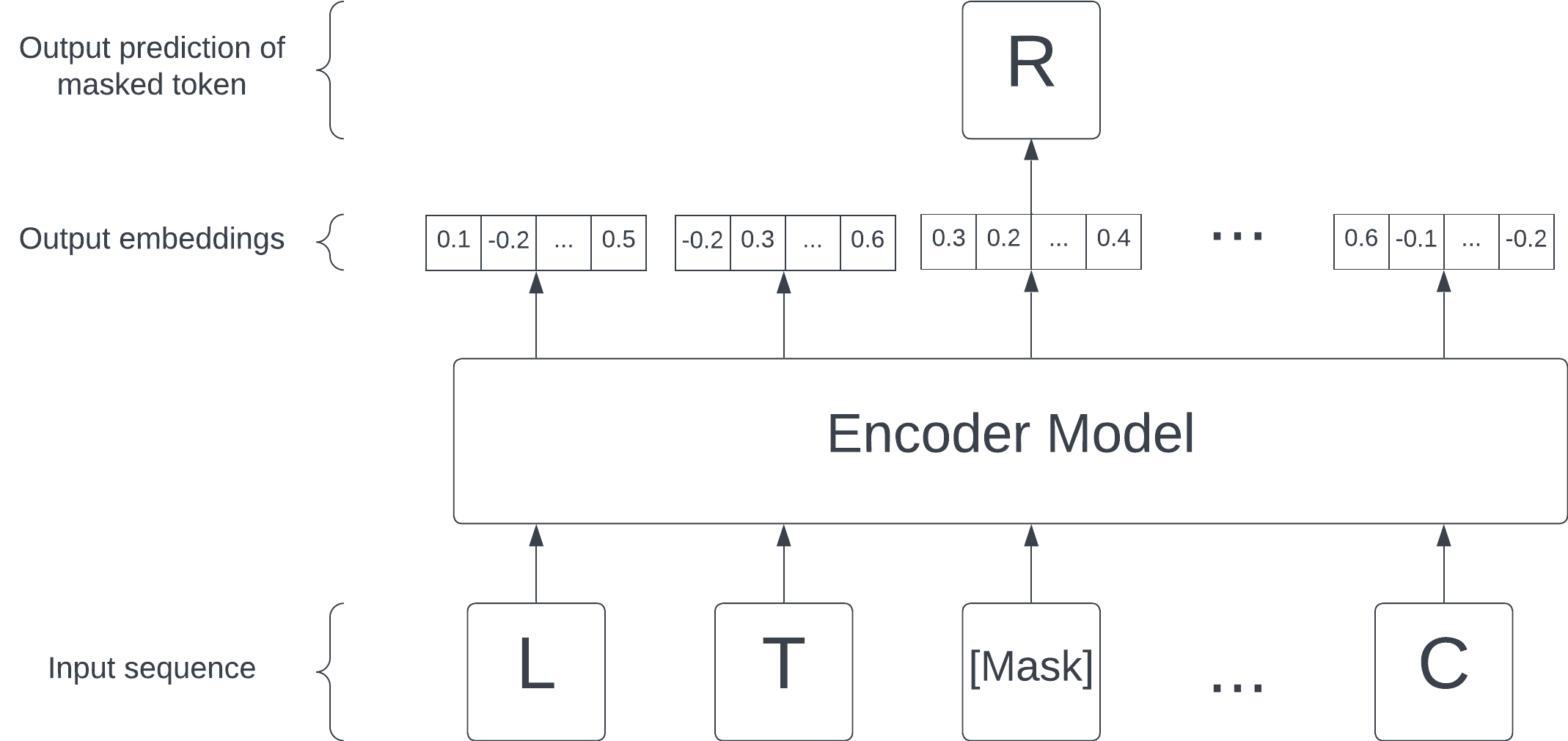
Though we will focus on masked language modeling in this article, another way to construct this self-supervision task is via causal language modeling: given some tokens, ask the model to predict the next one. This is the approach used in OpenAI's GPT.
The model
(This section requires some basic knowledge of deep learning. If you are new to deep learning, I can't recommend enough Andrej Karpathy's YouTube series on NLP, which starts from the foundations of deep learning.)
The first protein language encoder model of this kind is UniRep (universal representation), which used a technique called Long Short Term Memory (LSTM). (It uses the causal instead of masked language modelling objective, predicting amino acids from left to right.)
More recently, transformer models that rely on a mechanism called attention have taken the spotlight. BERT stands for Bidirectional Encoder Representations from Transformer and is a class of state-of-the-art natural language encoders developed at Google. We will talk in some detail about a BERT-like encoder model applied to proteins.
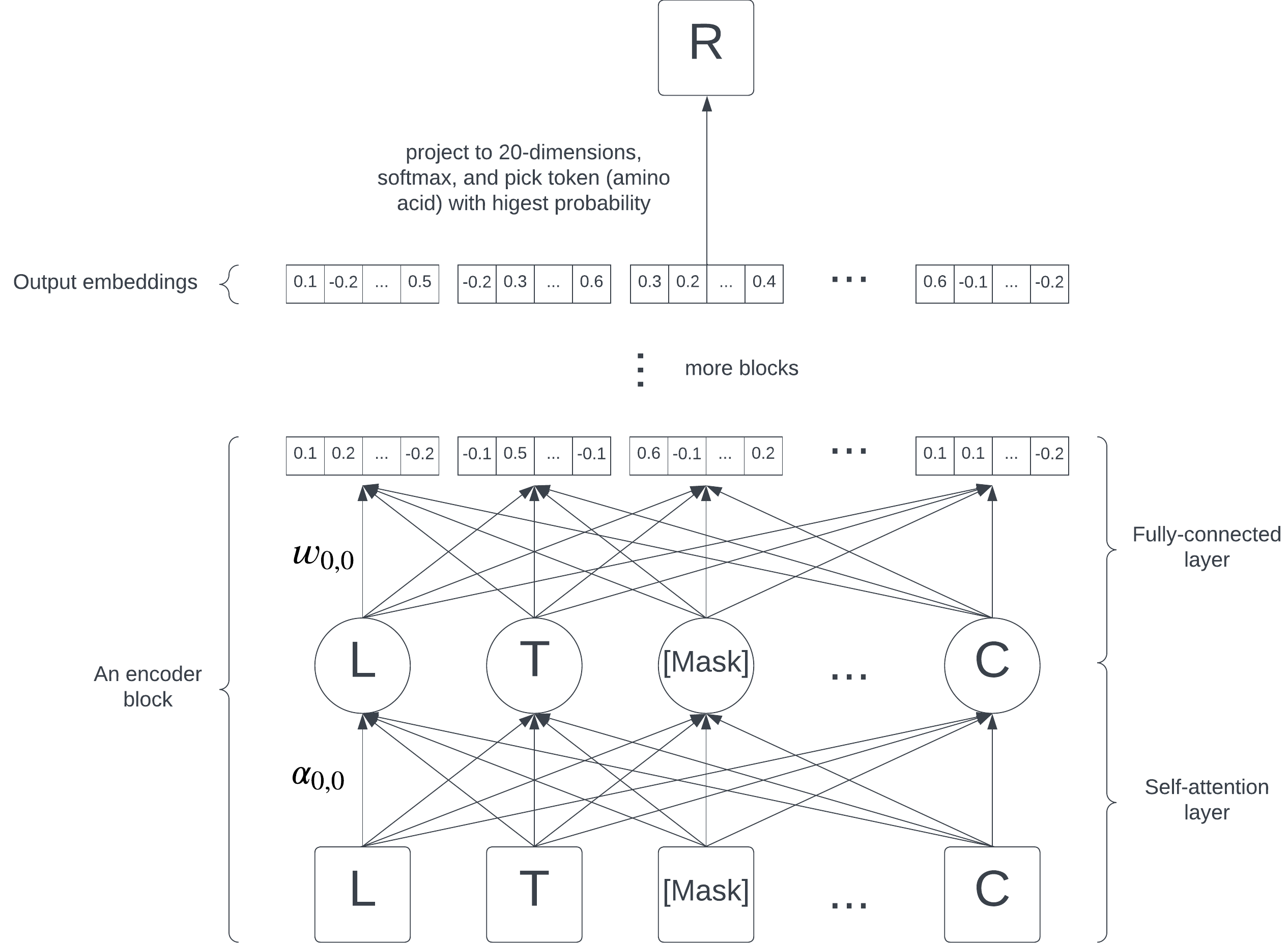
BERT consists of 12 encoder blocks, each containing a self-attention layer and a fully-connected layer. On the highest level, they are just a collection of numbers (parameters) learned by the model; each edge in the diagram represents a parameter.
Roughly speaking, the parameters in the self-attention layer, also known as attention scores, capture the alignment, or similarity, between two amino acids. If is large, we say that the token attends to the token. Intuitively, each token can attend to different parts of the sequence, focusing on what's relevant to it and glancing over what's not.
Here's an example of attention scores of a transformer trained on a word-tokenized sentence:
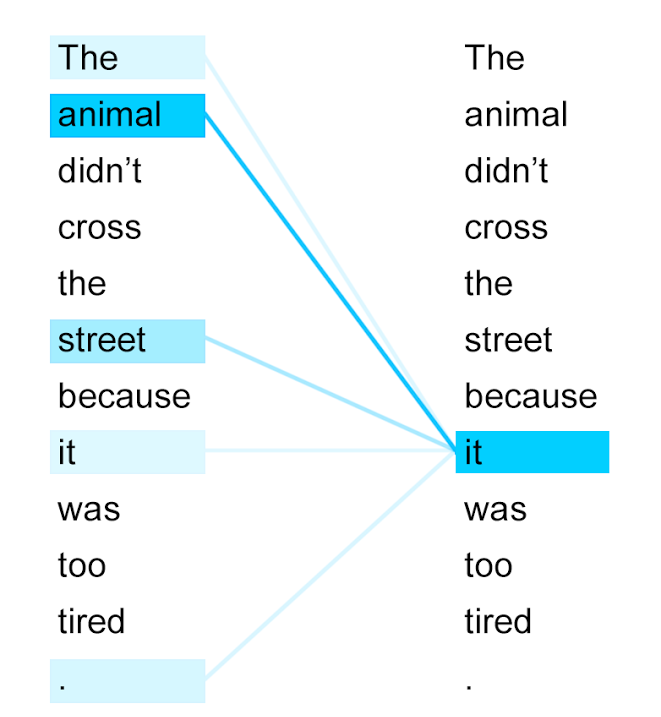
The token "it" attends strongly the token "animal" because of their close relationship – they refer to the same thing – whereas most other tokens are ignored. It is our goal to tease out similar semantic relationships between amino acids.
The details of how these attention scores are calculated are explained and visualized in Jay Alammar's amazing post The Illustrated Transformer. Here's a helpful explanation on how they differ from the weights in the fully-connected layer.
As it turns out, once we train our model on the masked language modelling objective, the output embeddings become rich representations of the underlying sequence (!) – what we have been looking for.